 Our laboratory pioneered the study of the oncogenic activity of G proteins and G protein coupled receptors (GPCRs), including virally-encoded GPCRs, and how their signaling circuitry regulates normal and aberrant cell growth and metastasis. We demonstrated the potent transforming activity of G proteins and GPCRs, including Gαq (GNAQ), Gα12 (GNA12) and their coupled GPCRs, and showed that this remarkable biological activity requires a novel signaling network involving mitogen activated kinases (MAPK), ERK, JNK, p38, and their regulation by small GTPases of the Ras and Rho family. The latter had broad implications in the signal transduction field, as we showed that Rho GTPases, which were known for their cytoskeletal effects, stimulate JNK, thereby providing the first demonstration that Rho proteins are integral components of signaling routes linking the cell surface to the nucleus. We also made the unexpected discovery that rather than G protein α subunits (Gα), Gβγ subunits initiate the activate ERK through Ras, thus providing the first demonstration that cell growth promotion by GPCRs involves a complex protein-protein interaction network rather than relying solely on diffusible second messengers. We also reported the first studies supporting that non-receptor tyrosine kinases and tyrosine kinase growth factor receptors converge with GPCRs in the activation of Rho-regulated MAPK signaling networks to control the expression of growth promoting genes.
Our laboratory pioneered the study of the oncogenic activity of G proteins and G protein coupled receptors (GPCRs), including virally-encoded GPCRs, and how their signaling circuitry regulates normal and aberrant cell growth and metastasis. We demonstrated the potent transforming activity of G proteins and GPCRs, including Gαq (GNAQ), Gα12 (GNA12) and their coupled GPCRs, and showed that this remarkable biological activity requires a novel signaling network involving mitogen activated kinases (MAPK), ERK, JNK, p38, and their regulation by small GTPases of the Ras and Rho family. The latter had broad implications in the signal transduction field, as we showed that Rho GTPases, which were known for their cytoskeletal effects, stimulate JNK, thereby providing the first demonstration that Rho proteins are integral components of signaling routes linking the cell surface to the nucleus. We also made the unexpected discovery that rather than G protein α subunits (Gα), Gβγ subunits initiate the activate ERK through Ras, thus providing the first demonstration that cell growth promotion by GPCRs involves a complex protein-protein interaction network rather than relying solely on diffusible second messengers. We also reported the first studies supporting that non-receptor tyrosine kinases and tyrosine kinase growth factor receptors converge with GPCRs in the activation of Rho-regulated MAPK signaling networks to control the expression of growth promoting genes.
Research
Our cancer research is focused on G proteins, GPCRs, HNSCC, and precision immunotherapy. Explore the panels below to learn more about our work.
G Proteins / GPCRS and Cancer
HNSCC / ORAL CANCER / PI3K-MTOR and Hippo Pathways
IMMUNE ONCOLOGY
UVEAL MELANOMA
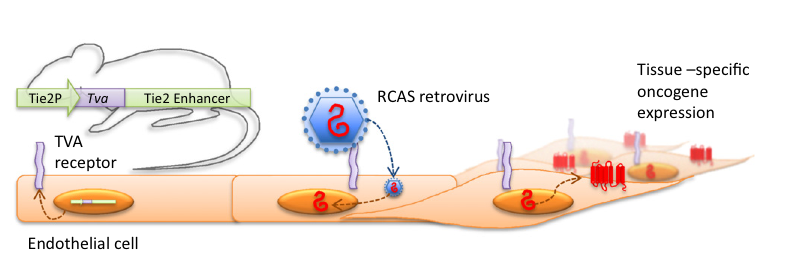 Our team revealed the transforming potential of a GPCR encoded by the open reading frame 74 of the Kaposi’s sarcoma (KS)-associated herpesvirus (KSHV) genome, thus providing a direct link between GPCRs and human viral-associated malignancies. For these studies, we developed a novel high throughput animal model system enabling the tissue-specific viral gene delivery and expression in mouse endothelial cells in vivo. This approach revealed that among all candidate KSHV oncogenes, only the viral GPCR was able to recapitulate KS sarcoma pathogenesis. Dissection of the underlying mechanism led to the discovery that the stimulation of AKT and mTOR by the viral GPCR were strictly necessary for sarcomagenesis. Our basic studies provided a rationale for the clinical evaluation by other groups of rapamycin, an mTOR inhibitor, in renal transplanted patients developing KS, which represented the first successful use of mTOR inhibiting agents to treat human malignancies. We also showed that the GPCR signaling specificity can be exploited by targeting selectively the PI3Kγ isoform for KS treatment, thereby circumventing the potential immunosuppressive activity of mTOR inhibitors in AIDS patients, who are at high risk of developing KS.
Our team revealed the transforming potential of a GPCR encoded by the open reading frame 74 of the Kaposi’s sarcoma (KS)-associated herpesvirus (KSHV) genome, thus providing a direct link between GPCRs and human viral-associated malignancies. For these studies, we developed a novel high throughput animal model system enabling the tissue-specific viral gene delivery and expression in mouse endothelial cells in vivo. This approach revealed that among all candidate KSHV oncogenes, only the viral GPCR was able to recapitulate KS sarcoma pathogenesis. Dissection of the underlying mechanism led to the discovery that the stimulation of AKT and mTOR by the viral GPCR were strictly necessary for sarcomagenesis. Our basic studies provided a rationale for the clinical evaluation by other groups of rapamycin, an mTOR inhibitor, in renal transplanted patients developing KS, which represented the first successful use of mTOR inhibiting agents to treat human malignancies. We also showed that the GPCR signaling specificity can be exploited by targeting selectively the PI3Kγ isoform for KS treatment, thereby circumventing the potential immunosuppressive activity of mTOR inhibitors in AIDS patients, who are at high risk of developing KS.
![]()
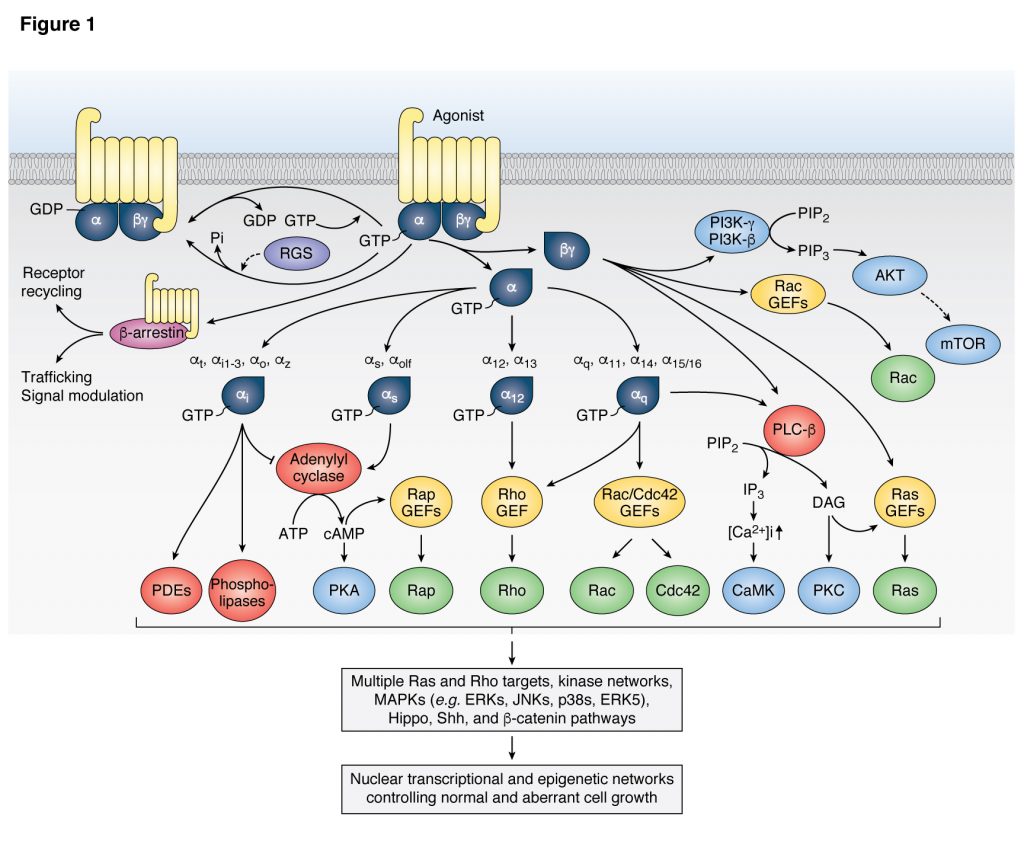
G protein-coupled receptors (GPCRs) represent the largest family of cell surface proteins involved in signal transmission. These receptors play key physiological roles, and their dysfunction contributes to some of the most prevalent human diseases. However, the contribution of GPCRs, their linked heterotrimeric G proteins, and their regulated signaling circuitries in cancer initiation, progression, metastatic spread, and cancer therapy resistance has not been thoroughly investigated, Emerging evidence from our laboratories at UCSD has revealed that malignant cells often hijack the normal physiological functions of GPCRs to proliferate autonomously, evade immune detection, enhance their nutrient and oxygen supply, invade their surrounding tissues, and disseminate to other organs. Strikingly, our recent analysis of human cancer genomes revealed an unanticipated high frequency of mutations in G proteins and GPCRs in most tumor types, which we refer to as the onco-GPCRome. Indeed, nearly 30% of human cancers harbor mutations in GPCRs or G proteins. For example, mutually exclusive activating mutations in GNAQ or GNA11 (encoding Gαq family members) occur in ~90% and ~4% of melanomas arising in the eye and skin, respectively, where they act as driver oncogenes. Remarkably, approximately 5% of all sequenced tumors harbor activated GNAS mutants, encoding a constitutively active Gαs. Oncogenic GNAS mutations were initially discovered in multiple endocrine tumors, such as growth hormone-secreting pituitary (28%) and thyroid adenomas (5%). However, we have recently found that GNAS is mutated in many human cancers, including colorectal carcinoma (CRC) (4%), pancreatic tumors (12%), hepatocellular carcinoma (2%) and, strikingly, appendix cancers (70%). In this regard, there is substantial epidemiological evidence supporting the efficacy of aspirin and other nonsteroidal anti-inflammatory drugs (NSAIDs) that block COX2 for the prevention of CRC. Our team discovered that prostaglandin E2 (PGE2), which accumulates due to COX2 overexpression, can act on CRC cells directly by stimulating the β-catenin pathway through Gs-linked PGE2 receptors, supporting the direct relevance of GNAS and its activated pathways for CRC initiation and progression. Overexpression of GPCRs and their activation by locally released and circulating agonists in an autocrine and paracrine fashion is also a frequent event in cancer. Overall, as GPCRs represent the target of >25% of all drugs in the market, we hypothesize that interfering with the human onco-GPCRome by targeting GPCRs, G proteins, or their downstream signaling pathways may provide a unique opportunity for the development of novel, mechanism-based strategies for cancer diagnosis, prevention and treatment.
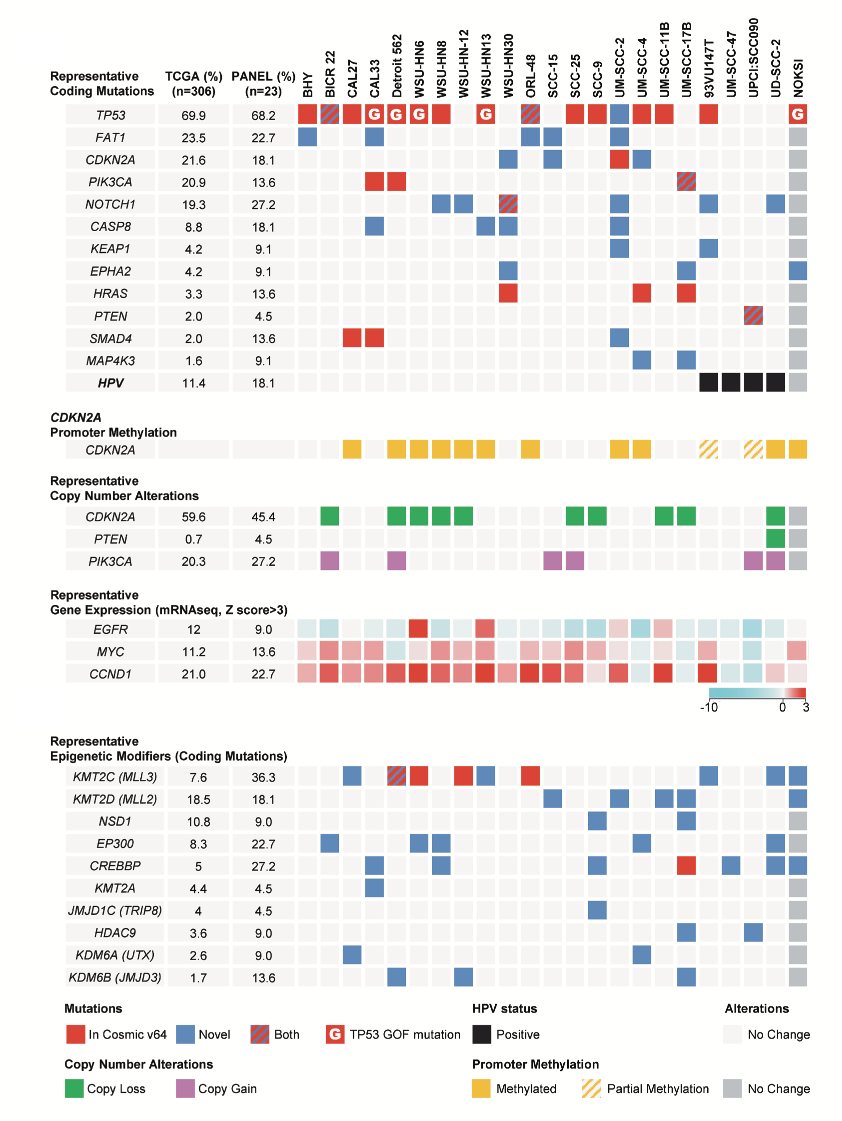 Based on our significant contributions to the dissection of growth promoting signaling networks, Dr. Gutkind was given the opportunity to develop and lead a new NIDCR and trans-NIH head and neck cancer (HNSCC) research program. Examples of the early success of our team science approach included the “Oral Cancer Genome Anatomy Project” a decade ago, and the “International Oral Cancer Tissue Array Initiative”, which provided a wealth of resources to the community and a platform for breakthrough discoveries moving the field forward. Among them, we focused on the molecular dissection of aberrant signaling networks in HNSCC. This led to our discovery that the persistent activation of the PI3K/mTOR signaling pathway is one of the most frequently dysregulated molecular mechanisms in HNSCC, including in HPV-associated lesions, and that mTOR inhibition causes tumor regression in a series of genetically-defined and chemically-induced experimental HNSCC models. These findings and the discovery that most HNSCCs exhibit genomic alterations leading to PI3K/mTOR activation, provided a strong rationale for launching a multi-institutional clinical trial (NCT01195922), in which Dr. Gutkind serve as the PI, inhibiting mTOR with rapamycin in newly diagnosed HNSCC patients. The trial has been recently completed with encouraging results in terms of objective responses and limited toxicities.
Based on our significant contributions to the dissection of growth promoting signaling networks, Dr. Gutkind was given the opportunity to develop and lead a new NIDCR and trans-NIH head and neck cancer (HNSCC) research program. Examples of the early success of our team science approach included the “Oral Cancer Genome Anatomy Project” a decade ago, and the “International Oral Cancer Tissue Array Initiative”, which provided a wealth of resources to the community and a platform for breakthrough discoveries moving the field forward. Among them, we focused on the molecular dissection of aberrant signaling networks in HNSCC. This led to our discovery that the persistent activation of the PI3K/mTOR signaling pathway is one of the most frequently dysregulated molecular mechanisms in HNSCC, including in HPV-associated lesions, and that mTOR inhibition causes tumor regression in a series of genetically-defined and chemically-induced experimental HNSCC models. These findings and the discovery that most HNSCCs exhibit genomic alterations leading to PI3K/mTOR activation, provided a strong rationale for launching a multi-institutional clinical trial (NCT01195922), in which Dr. Gutkind serve as the PI, inhibiting mTOR with rapamycin in newly diagnosed HNSCC patients. The trial has been recently completed with encouraging results in terms of objective responses and limited toxicities.
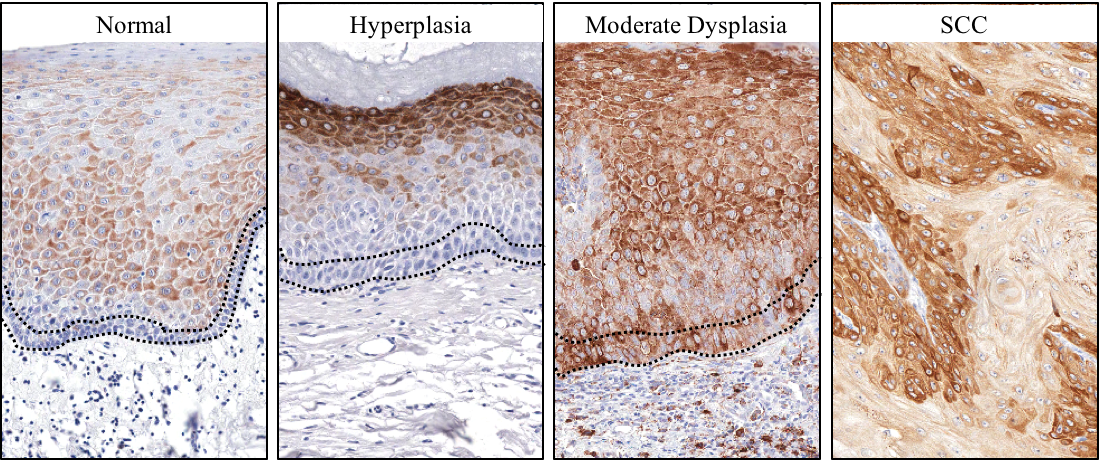
There is an urgent need to develop prevention options to halt the progression of oral premalignant lesions (OPL) into OSCC. Our lab discovered that activation of mTOR is an early event in OPL, and we showed that metformin, which is currently used by millions of Americans for type 2 diabetes, inhibits mTOR indirectly in OPL and their potential cancer initiating cells, thereby halting tumor progression. Following our passion for translating lab’s findings into effective cancer therapies, we relocated our research team to the UCSD Moores Cancer Center. Based on our discoveries, and recent epidemiological data showing a significant reduction in OSCC incidence in diabetic patients on metformin, our multidisciplinary team launched a multi institutional phase IIa clinical trial (NCT02581137) to explore the potential use of metformin for OSCC prevention. The results from our clinical trial were remarkable, with 60% of the patients displaying favorable histological responses and ~20% complete responses after three months of metformin administration. Based on the positive results we have recently launched double blind, randomized, placebo-controlled Phase IIB trial, including 10 centers in the United States and Canada (NCT05237960). This ongoing trial may provide a precision prevention strategy for OSCC, and reveal that the use of metformin may represent a potential safe and low cost chemopreventive approach for OSCC in at risk patients.
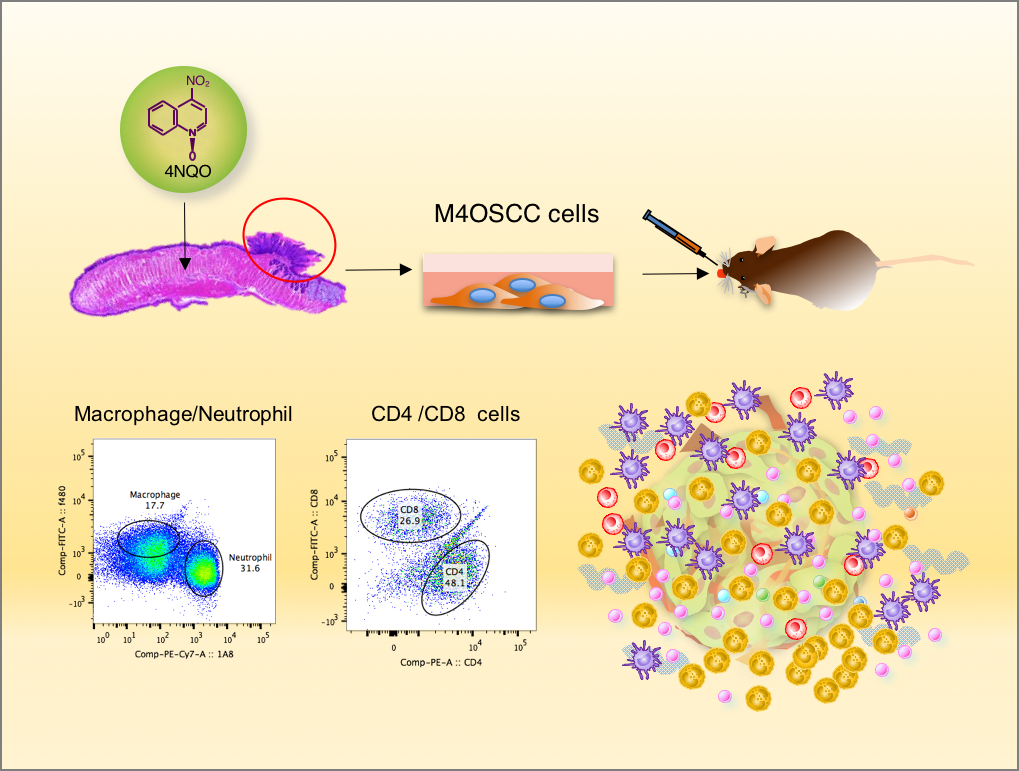
For patients with recurrent or metastatic HNSCC, standard of care combination chemotherapy yields a median overall survival of only 10 months and long-term survival is rare. Most tumors deploy multiple mechanisms to avoid immune recognition and an anti-tumor immune response. A key emerging mechanism of tumor immunosuppression involves T cell exhaustion, whereby T cell reactivity is impaired due to activation of T cell checkpoints, including PD-1, by its ligand, PD-L1 that is expressed by multiple immune cells and some cancer cells, including HNSCC. Recent revolutionary therapeutic strategies restoring T cell mediated anti-tumor immunity by immune oncology (IO) agents targeting immune checkpoints, including CTLA-4, PD-1, and PDL-1, have achieved remarkable clinical responses. In HNSCC, anti-PD-1 antibodies (nivolumab and pembrolizumab) demonstrated immune modulation and durable remissions, but the overall response rate was only 20% and only ~30% of patients were alive one year after the initiation of therapy.
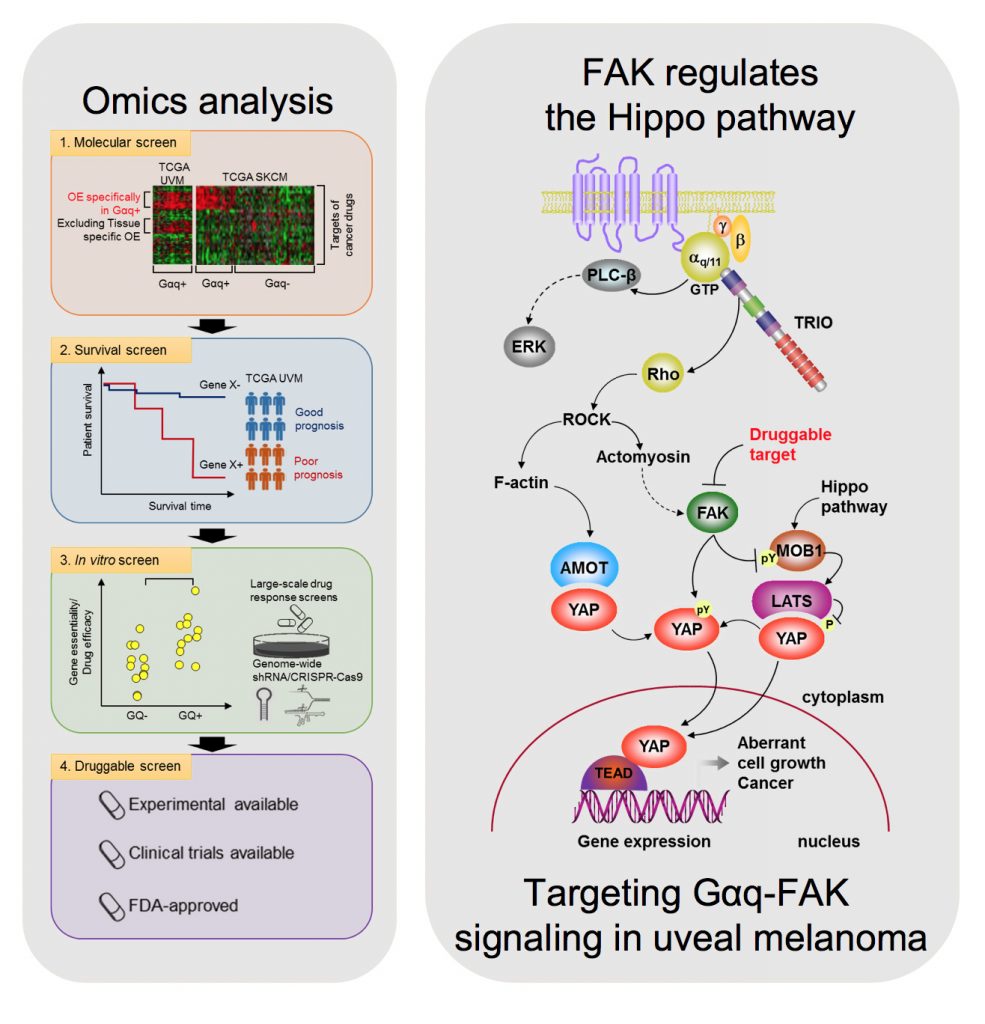
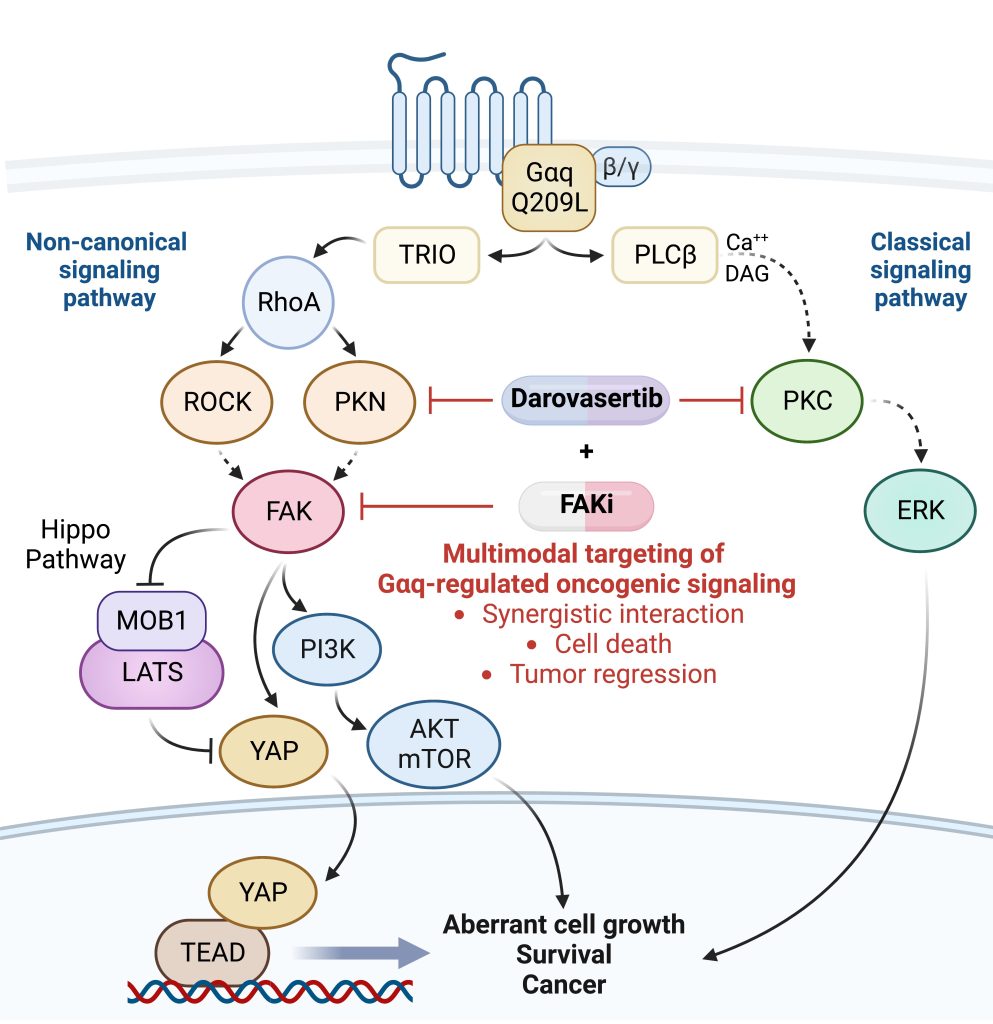
Uveal melanoma (UM) is the most common primary cancer of the eye in adults, and is the second most common melanoma subtype after skin cutaneous melanoma (SKCM). UM is diagnosed in about 2,500 patients each year in the United States alone, nearly 50% of which will die from liver metastasis within 5-10 years after diagnosis, independent of the successful treatment of the primary lesions. Activating mutations in GNAQ and GNA11 (often referred to as GNAQ oncogenes, which encode GTPase deficient and constitutively active Gαq proteins), were identified in ~93% of UM and 4% of SKCM, respectively, where they act as oncogenic driver oncogenes. These findings and our team’s early discovery that activated mutants of Gαq represent a new class of oncogenes firmly established UM and a subset of SKCM as Gαq-driven human malignancies. Indeed, this provides a clear example of a human malignancy that is initiated by gain of function mutations in Gαq and Gα11 proteins. The best-known downstream signaling event initiated by Gαq involves its ability is to activate phospholipase C (PLC) β and the consequent increased hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to produce two second messengers: inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 raises cytoplasmic Ca2+ levels, which stimulates multiple calcium-regulated pathways and, together with DAG, activates classic protein kinase C (PKC) isoforms. However, the molecular events underlying GNAQ-driven malignancies are not yet defined, thus limiting the ability to develop novel anticancer-targeted therapies. In our recent studies, we found that activating mutation of Gαq can trigger YAP translocation into the nucleus and stimulates YAP-dependent transcription, and that this process is independent from PLCβ stimulation but requires the activation of a Gαq-regulated guanine nucleotide exchange factor, TRIO, and the subsequent activation of the small GTPases RhoA and Rac1 and their associated signaling networks. Gαq activation is sufficient to stimulate YAP in animal cancer models, and knock down of Gαq in uveal melanomas harboring GNAQ mutations reduces the nuclear location of YAP, and YAP-dependent gene transcription. Gαq-TRIO-Rho/Rac signaling circuitry contributes to YAP-dependent growth in uveal melanoma, which is the first described GNAQ/GNA11-initiated human malignancy, and, thus, that YAP may represent a novel therapeutic target for uveal melanoma treatment.
However, there are currently limited option to target YAP therapeutically. Thus, there is an urgent need to understand how Gαq promotes cancer growth in order to develop new targeted (precision) therapeutic options. In a recent study, we used a novel computational framework to shed light on Gαq biology and identify systems vulnerabilities for UM, based on the prediction of synthetic (dosage) lethal gene interactions of Gαq. This novel pipeline integrates data from large multiomics cancer datasets, including UM patient transcriptomes and genomes, with in vitro screens (datasets of gene essentiality and dependence and druggable screens (datasets of drug response screens). The top predicted synthetic lethal gene with GNAQ was PTK2, suggesting the potential benefit of targeting the PTK2 gene product (a non-receptor tyrosine kinase known as FAK) in UM. Remarkably, activation of FAK by Gq-linked receptors was initially reported by our team in the early 90s. This unexpected convergence of computational predictions, biochemical, and genetic information prompted us to focus on the role of this Gαq-tyrosine kinase signaling axis in UM. We found that gene editing or inhibition of FAK reduces UM cell growth and tumor formation in vivo. Furthermore, systematic dissection of the underlying mechanisms led us to uncover that FAK promotes YAP activity through direct tyrosine phosphorylation of YAP concomitant with the release of inhibitory core Hippo signaling on YAP. Our studies provided a novel direct link between Gαq-FAK driven tyrosine phosphorylation networks and YAP activation. Based on these findings, the use of FAKi for the treatment of metastatic UM is under current evaluation, as a single agent and as part of a signal-transduction based multimodal therapy.
As part of these studies, we have used unbiased genetic screens to identify new treatment options for UM. We discovered that inhibition of the non-receptor tyrosine kinase FAK and the adaptive activation of the ERK pathway downstream from GNAQ results in UM cell death and regression of liver metastatic lesions. This provided the foundation for the first signal transduction-based multimodal precision therapy in metastatic UM (mUM)(NCT04720417). Our recent large chemogenetic drug screen revealed that targeting FAK and PKC/PKN represents an effective precision-guided combination, which we plan to explore in the clinic shortly.